[1]:
import pandas as pd
from nl_causal.ts_models import _2SLS, _2SIR
from nl_causal.linear_reg import L0_IC
import numpy as np
from sklearn.preprocessing import normalize
from sim_data import sim
from sklearn.preprocessing import StandardScaler
from scipy import stats
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import power_transform, quantile_transform
import random
from os import listdir
from os.path import isfile, join, isdir
import statsmodels.api as sm
from sklearn.feature_selection import VarianceThreshold
from os import path
[2]:
mypath = 'path_to_data'
gene_folders = [name for name in listdir(mypath) if isdir(join(mypath, name))]
[3]:
np.random.seed(0)
np.set_printoptions(formatter={'float': lambda x: "{0:0.4f}".format(x)})
df = {'gene': [], 'p-value': [], 'beta': [], 'method': []}
[ ]:
for folder_tmp in gene_folders:
if 'July20_2SIR' not in folder_tmp:
continue
gene_code = folder_tmp.replace('July20_2SIR', '')
print('\n##### Causal inference of %s #####' %gene_code)
## load data
dir_name = mypath+'/'+folder_tmp
sum_stat = pd.read_csv(dir_name+"/sum_stat.csv", sep=' ', index_col=0)
gene_exp = -pd.read_csv(dir_name+"/gene_exp.csv", sep=' ', index_col=0)
snp = pd.read_csv(dir_name+"/snp.csv", sep=' ', index_col=0)
## exclude the gene with nan in the dataset
if sum_stat.isnull().sum().sum() + snp.isnull().sum().sum() + gene_exp.isnull().sum().sum() > 0:
continue
if not all(sum_stat.index == snp.columns):
print('The cols in sum_stat are not corresponding to snps, we rename the sum_stat!')
sum_stat.index = snp.columns
## n1 and n2 is pre-given
n1, n2, p = len(gene_exp), 54162, snp.shape[1]
LD_Z1, cov_ZX1 = np.dot(snp.values.T, snp.values), np.dot(snp.values.T, gene_exp.values.flatten())
LD_Z2, cov_ZY2 = LD_Z1/n1*n2, sum_stat.values.flatten()*n2
## 2SLS
LS = _2SLS()
## Stage-1 fit theta
LS.fit_theta(LD_Z1, cov_ZX1)
## Stage-2 fit beta
LS.fit_beta(LD_Z2, cov_ZY2, n2)
## produce p_value for beta
LS.test_effect(n2, LD_Z2, cov_ZY2)
print('-'*20)
print('LS beta: %.3f' %LS.beta)
print('p-value based on 2SLS: %.5f' %LS.p_value)
## compute R2 for the 2SLS
## save the record
df['gene'].append(gene_code)
df['method'].append('2SLS')
df['p-value'].append(LS.p_value)
df['beta'].append(LS.beta)
## PT-2SLS
PT_X1 = power_transform(gene_exp.values.reshape(-1,1), method='yeo-johnson').flatten()
PT_X1 = PT_X1 - PT_X1.mean()
PT_cor_ZX1 = np.dot(snp.values.T, PT_X1)
PT_LS = _2SLS()
## Stage-1 fit theta
PT_LS.fit_theta(LD_Z1, PT_cor_ZX1)
## Stage-2 fit beta
PT_LS.fit_beta(LD_Z2, cov_ZY2, n2)
## produce p-value for beta
PT_LS.test_effect(n2, LD_Z2, cov_ZY2)
print('-'*20)
print('PT-LS beta: %.3f' %PT_LS.beta)
print('p-value based on PT-2SLS: %.5f' %PT_LS.p_value)
## save the record
df['gene'].append(gene_code)
df['method'].append('PT-2SLS')
df['p-value'].append(PT_LS.p_value)
df['beta'].append(PT_LS.beta)
## 2SIR
SIR = _2SIR(data_in_slice=0.2*n1)
## Stage-1 fit theta
SIR.fit_theta(Z1=snp.values, X1=gene_exp.values.flatten())
## Stage-2 fit beta
SIR.fit_beta(LD_Z2, cov_ZY2, n2)
## generate p-value for beta
SIR.test_effect(n2, LD_Z2, cov_ZY2)
print('-'*20)
print('2SIR eigenvalues: %.3f' %SIR.sir.eigenvalues_)
print('2SIR beta: %.3f' %SIR.beta)
print('p-value based on 2SIR: %.5f' %SIR.p_value)
## save the record
df['gene'].append(gene_code)
df['method'].append('2SIR')
df['p-value'].append(SIR.p_value)
df['beta'].append(SIR.beta)
## Comb-2SIR
data_in_slice_lst = [.05*n1, .1*n1, .2*n1, .3*n1, .5*n1]
comb_pvalue, comb_beta, comb_eigenvalue = [], [], []
for data_in_slice_tmp in data_in_slice_lst:
SIR = _2SIR(data_in_slice=data_in_slice_tmp)
## Stage-1 fit theta
SIR.fit_theta(Z1=snp.values, X1=gene_exp.values.flatten())
## Stage-2 fit beta
SIR.fit_beta(LD_Z2, cov_ZY2, n2)
## generate p-value for beta
SIR.test_effect(n2, LD_Z2, cov_ZY2)
comb_beta.append(SIR.beta)
comb_pvalue.append(SIR.p_value)
comb_eigenvalue.append(SIR.sir.eigenvalues_[0])
comb_T = np.tan((0.5 - np.array(comb_pvalue))*np.pi).mean()
correct_pvalue = min( max(.5 - np.arctan(comb_T)/np.pi, np.finfo(np.float64).eps), 1.0)
print('-'*20)
print('Comb-2SIR eigenvalues: %.3f' %np.mean(comb_eigenvalue))
print('Comb-2SIR beta: %.3f' %np.mean(comb_beta))
print('p-value based on Comb-2SIR: %.5f' %correct_pvalue)
## save the record
df['gene'].append(gene_code)
df['method'].append('Comb-2SIR')
df['p-value'].append(correct_pvalue)
df['beta'].append(np.mean(comb_beta))
df = pd.DataFrame.from_dict(df)
[5]:
df.sample(10)
[5]:
| gene | p-value | beta | method | |
|---|---|---|---|---|
| 21690 | CLPS | 0.987946 | 0.000188 | 2SIR |
| 39533 | IFNA10 | 0.827260 | -0.002878 | PT-2SLS |
| 15321 | RASSF2 | 0.763675 | 0.003542 | PT-2SLS |
| 27167 | USP3 | 0.106309 | 0.018060 | Comb-2SIR |
| 39426 | SLC25A51 | 0.658329 | 0.005132 | 2SIR |
| 38990 | HLF | 0.186863 | 0.014551 | 2SIR |
| 1322 | MASP2 | 0.377253 | 0.011340 | 2SIR |
| 46601 | SCARA5 | 0.149602 | -0.015242 | PT-2SLS |
| 57909 | EFTUD1 | 0.039076 | -0.029672 | PT-2SLS |
| 635 | MSL1 | 0.315792 | 0.013196 | Comb-2SIR |
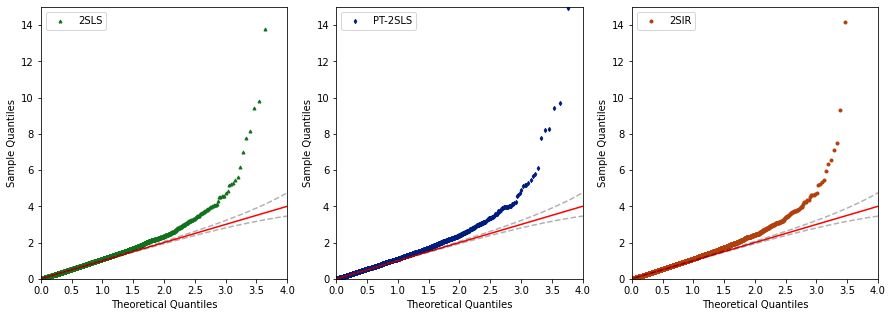
QQ-plot for p-values
[7]:
import matplotlib.pyplot as plt
import scipy.stats as stats
import seaborn as sns
from scipy.stats import rv_continuous
import statsmodels.api as sm
import random
from scipy.stats import beta, chi2
from nl_causal.base.rv import neg_log_uniform
[8]:
ci = 0.95
methods = ['2SLS', 'PT-2SLS', '2SIR']
colors = [sns.color_palette("dark")[2], sns.color_palette("dark")[0], sns.color_palette("dark")[1]]
[9]:
all_genes = list(set(df['gene']))
num_gene = len(all_genes)
## points for CIs
low_bound = [beta.ppf((1-ci)/2, a=i, b=num_gene-i+1) for i in range(1,num_gene+1)]
up_bound = [beta.ppf((1+ci)/2, a=i, b=num_gene-i+1) for i in range(1,num_gene+1)]
ep_points = (np.arange(1,num_gene+1) - 1/2) / num_gene
[11]:
fig, axs = plt.subplots(1,3,figsize=(15, 5))
for i in range(3):
sm.qqplot(-np.log10(df[df['method'] == methods[i]]['p-value'].values), dist=neg_log_uniform(), line="45", ax=axs[i])
axs[i].set_xlim([0,4.0])
axs[i].set_ylim([0,15])
axs[i].get_lines()[0].set_markersize(3.0)
axs[i].get_lines()[0].set_markeredgecolor(colors[i])
axs[i].get_lines()[0].set_markerfacecolor(colors[i])
axs[i].get_lines()[0].set_markerfacecoloralt(colors[i])
axs[0].get_lines()[0].set_marker('^')
axs[1].get_lines()[0].set_marker('d')
axs[2].get_lines()[0].set_marker('o')
axs[0].get_lines()[0].set(label='2SLS')
axs[1].get_lines()[0].set(label='PT-2SLS')
axs[2].get_lines()[0].set(label='2SIR')
for i in range(3):
axs[i].plot(-np.log10(ep_points), -np.log10(low_bound), 'k--', alpha=0.3)
axs[i].plot(-np.log10(ep_points), -np.log10(up_bound), 'k--', alpha=0.3)
axs[i].legend()
plt.show()
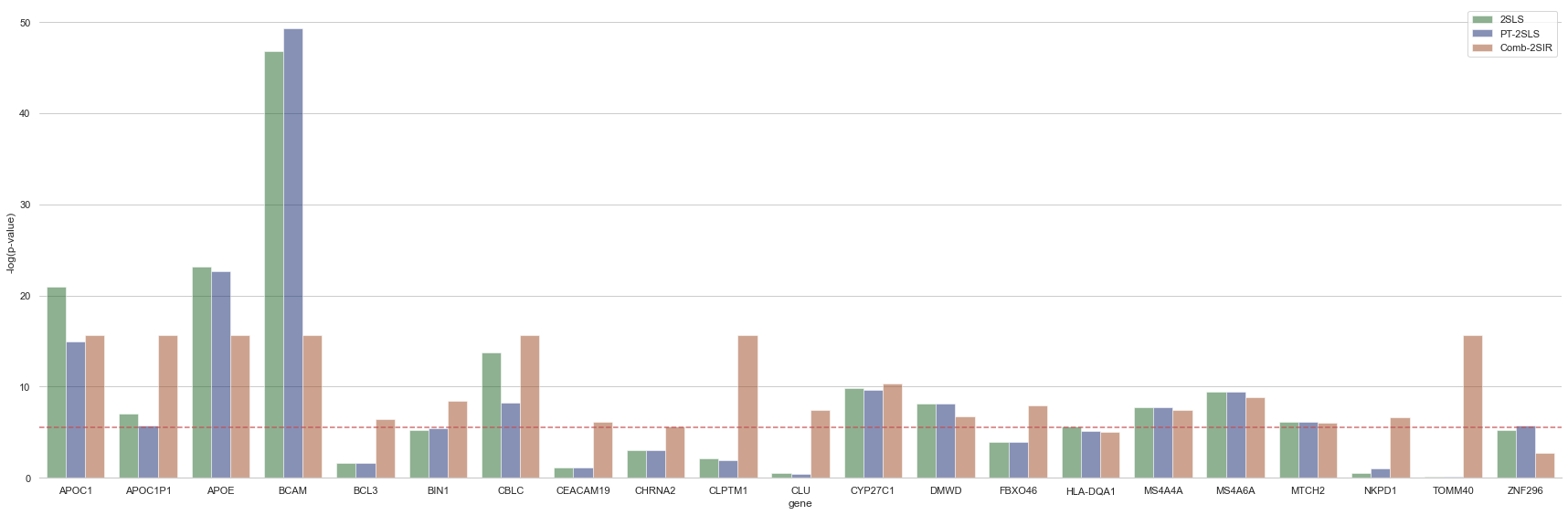
Bar-plot for significant genes
[12]:
import seaborn as sns
import matplotlib.pyplot as plt
[13]:
gene_set = list(set(df['gene']))
num_gen = len(gene_set)
level = 0.05 / num_gen
[14]:
# take the gene with at least one siginificant p-value
min_p = df.groupby('gene')['p-value'].min()
gene_set = list(min_p[min_p < level].index)
[15]:
df = df[df['gene'].isin(gene_set)]
df['log-p-value'] = - np.log10( df['p-value'] )
## we only print the results for comb-2SIR
df = df[df['method'] != '2SIR']
[16]:
gene_set = set(df['gene'])
gene_set = list(gene_set)
gene_set.sort()
## plot for the final results
sns.set_theme(style="whitegrid")
# Draw a nested barplot by species and sex
plt.rcParams["figure.figsize"] = (16,6)
g = sns.catplot(
data=df, kind="bar", order=gene_set,
x="gene", y="log-p-value", hue="method",
alpha=.5, height=8, legend=False, aspect=3,
# palette='deep'
# palette=sns.color_palette(['green', 'blue', 'orange'])
palette = [sns.color_palette("dark")[2], sns.color_palette("dark")[0], sns.color_palette("dark")[1]]
)
plt.axhline(-np.log10(level), ls='--', color='r', alpha=.8)
g.despine(left=True)
g.set_axis_labels("gene", "-log(p-value)")
plt.legend(loc='upper right')
plt.show()